1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
| BiocManager::install(clusterProfiler) #使用富集分析會用到的package
library(clusterProfiler)
BiocManager::install(fgsea)
library(readxl) #可以讀入excel的package
# 以這組基因列表當練習: https://drive.google.com/open?id=1KTrQqok9cm5kPneo-Z8dWGEHaCNseUjp
#引用來源: https://www.sciencedirect.com/science/article/pii/S0306456518302456?via%3Dihub#f0020
#由於該文獻只有提供GI number,所以用DAVID的Gene ID Conversion Tool轉換成ENTREZ,不過原本有103個差異基因,轉換後只對的到70個
GO<-read_xlsx('GO.xlsx',col_names = T) #輸入資料,差異表達的基因 (ENTREZID命名格式)
BiocManager::install(enrichGO)
library(DOSE)
GO_BP <- enrichGO(gene =as.character(GO$ENTREZ_GENE_ID), #輸入的基因列表需轉換成字元
OrgDb = org_gal, #物種註釋數據庫,使用NCBI的database
keyType = 'ENTREZID', #gene命名格式
ont= 'BP', #選擇基因功能: 是BP (Biological Process), CC (Cellular Component), MF (Molecular Function)
pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
minGSSize = 10, #每個基因集最小數目
maxGSSize = 500, #用於測試的基因註釋最大數目
readable = FALSE,
pool = FALSE)
write.csv(summary(GO_BP),'GO_BP.csv',row.names =FALSE) #寫出GO分類表格
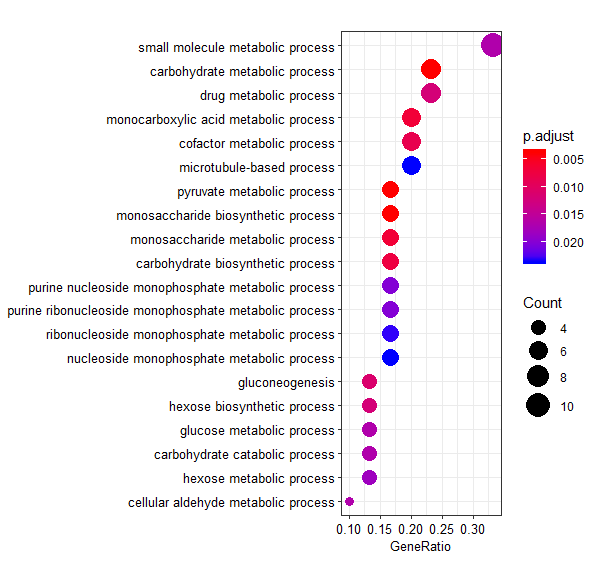
dotplot(GO_BP,x= 'GeneRatio',font.size= 10,color ='p.adjust',showCategory = 20) #泡泡圖, x可以改成以count表示,font.size: 文字大小; showCategory: 顯示的分類數,從p值最小的開始顯示。其他像是格式或是圖形樣式也可以變換,但我還沒摸熟。
|