
研究雞的腸道菌相除了要做基礎研究外,大多是為了提升生長和生產表現以及減少對抗生素依賴
來看看近幾年的相關研究的菌相分析方法以及分析流程會使用到哪些工具

Fig. 1. Overview of the factors affecting chicken health, welfare and performance and future perspectives in the analysis of the chicken microbiome (Borda-Molina et. al.,2018).
分析方法統整
下表整理了4篇文獻分析流程使用到的工具
| Huang et. al., 2018 | Segura-Wang et. al., 2021 | Feng et. al, 2021 | Temmerman et. al., 2022 | |
|---|---|---|---|---|
| Data availability | PRJNA417359 | PRJNA715658 | 4 bioprojects | PRJNA761967 |
| Platform | Illumina HiSeq 2500 & HiSeq X10 | Illumina NextSeq 500 V2 | Illumina | Illumina NextSeq 500 |
| Quality control | trimmomatic | fastp,Cutadapt | trimmomatic | |
| Remove reads of host | bwa-mem | DeconSeq | BMTagger | |
| Assembly | MegaHit | idba_ud | MegaHit | |
| Binning | MetaBAT2 | MetaWRAP | ||
| Co-assembly | MegaHit | SPAdes | ||
| Completeness | CheckM | |||
| Taxonomic classification | CARMA3 | GTDB-Tk | GTDB-TK,Kraken2 | Kraken2 |
| Taxonomic abundance | bwa-mem | Bracken | Bracken | |
| Phylogenetic tree | PhyloPhlAn,iTOL | |||
| Differential abundance | Kruskal-Wallis | DESeq2, Wald test | DESeq2, Mann–Whitney test | |
| biodiversity | Phyloseq (Bray-Curtis) | Vegan (Shannon,Chao1) | Vegan (Shannon,Bray–Curtis) | Vegan (Simpson,Bray–Curtis) |
| Gene prediction | Prodigal | Prodigal | ||
| Gene catalog | cd-hit-est | MMseq2 | ||
| Functional annotation | diamond (KEGG, eggNOG) | RAST,dbCAN2 | eggNOG-mapper,KofamKOALA | |
| ARG | diamond (CARD) | bwa-mem2 against ResFinder | ResFinder,ABRicate | usearch against MEGARes v2.0 |
| horizontal gene transfer | MetaCHIP |
4 bioprojects: PRJEB33338,PRJEB22062,PRJNA417359,PRJNA408020
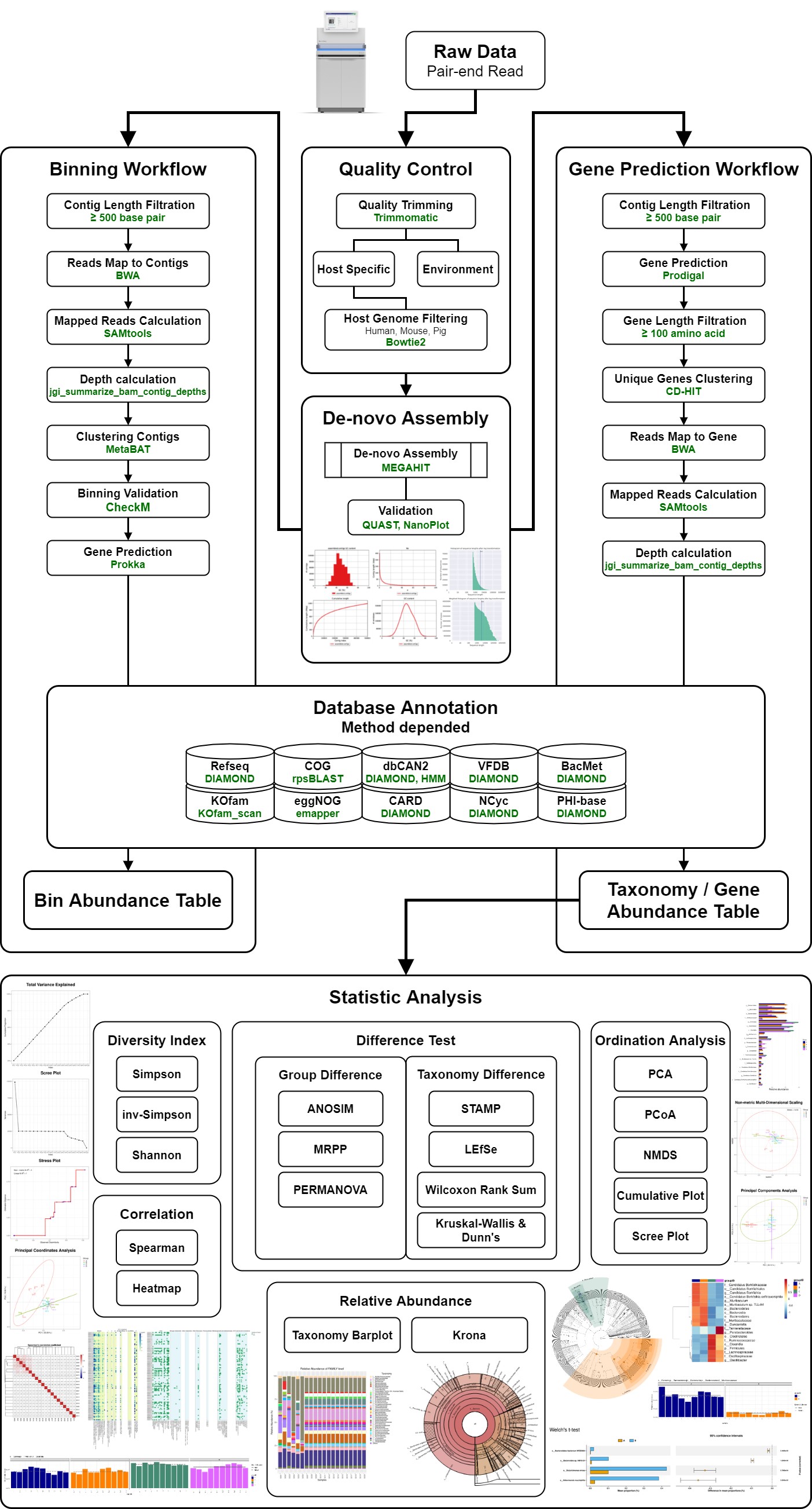
可以參考圖爾思的NGS metagenome分析流程了解菌相分析會做哪些分析處理
他們的流程圖已經幾乎把metagenome可以玩的分析方法都呈現出來了
References
- Huang, P., Zhang, Y., Xiao, K., Jiang, F., Wang, H., Tang, D., Liu, D., Liu, B., Liu, Y., He, X., Liu, H., Liu, X., Qing, Z., Liu, C., Huang, J., Ren, Y., Yun, L., Yin, L., Lin, Q., Zeng, C., … Zeng, J. (2018). The chicken gut metagenome and the modulatory effects of plant-derived benzylisoquinoline alkaloids. Microbiome, 6(1), 211. https://doi.org/10.1186/s40168-018-0590-5
- Segura-Wang, M., Grabner, N., Koestelbauer, A., Klose, V., & Ghanbari, M. (2021). Genome-Resolved Metagenomics of the Chicken Gut Microbiome. Frontiers in microbiology, 12, 726923. https://doi.org/10.3389/fmicb.2021.726923
- Feng, Y., Wang, Y., Zhu, B., Gao, G. F., Guo, Y., & Hu, Y. (2021). Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Communications biology, 4(1), 1305. https://doi.org/10.1038/s42003-021-02827-2
- Temmerman, R., Ghanbari, M., Antonissen, G., Schatzmayr, G., Duchateau, L., Haesebrouck, F., Garmyn, A., & Devreese, M. (2022). Dose-dependent impact of enrofloxacin on broiler chicken gut resistome is mitigated by synbiotic application. Frontiers in microbiology, 13, 869538. https://doi.org/10.3389/fmicb.2022.869538
問題與討論
提出一些我覺得比較新穎或疑惑的地方出來討論
Removal of contaminome
(Huang et. al., 2018)除了去除host genome還有food (maize, soybean, wheat, and zebrafish),大多文獻只會去除host genome
的確吃下去的飼糧也是個汙染源,但推測汙染的佔比應該很小
植物因為細胞壁的關係要萃取出DNA效率相較微生物DNA要低很多
如果在採樣前又斷食的話那來自飼糧的汙染佔比又會更低
但在科研角度這是很棒的做法
只可惜他沒有探討來自飼糧汙染的佔比有多高,否則會更有說服力
Abundance計算方法
Abundance的計算方式還是滿疑惑的,paper幾乎都不會交代完整詳細的計算過程
可能是就照主流怎麼算就怎麼算吧
常看到的有3種abundance
- Taxonomy
- MAG
- Gene
不管是哪一種源頭都是要從mapped reads來計算
計算方式都應該是參考RNA-seq的校正方法與計量公式
傳統通常會用TPM或FPKM,雖然這些方法都有誤差問題
但這以兩個公式較容易解釋,原理都是以gene length校正read count
換到metagnome分析就是以genome length校正
跟我以前看環境樣本metagenome的文獻一樣,幾乎沒有人會談到genome length校正問題
好在還是有看到有發表在Nature上的paper有提到以read count計算abundance需要以genome length校正
The resulting read abundance distributions require subsequent normalization by genome length (Milanese et. al., 2019).
但問題來了,metagenome不像RNA-seq幾乎所有的mRNA都是已知且是單一物種
尤其是環境樣本的metagenome可能九成以上都是未知的genome
而這些genome的genome length當然也是未知的
所以只要看到abundance的圓餅圖有unclassified表示它直接用mapped reads計算
或是給unclassified reads一個推測的數值
至於沒有unclassified的圓餅圖十之八九是先把unclassified reads排除之後再計算abundance
這種就只能呈現出已知的物種,如果未知的物種佔比越高對於結果呈現的偏差就會越大
因為看到的高豐度的物種可能佔整體的metagenome只是一小部分
但因為排除unclassified reads因而誤判它是優勢菌種
既然有未知菌種且無法得知其genome length,有的研究索性就不校正直接以read count計算abundance
雖然不能直接肯定,但Huang et. al., 2018應該是忽略物種豐度計算問題,改而只探討基因豐度
但其實基因豐度應該是不能只用gene catalog為母體,那也只是從有組裝出的MAG得來的而已
沒有被組裝出來的MAG絕對很多,只要是沒辦法貼到MAGs上的reads都是屬於沒有組裝出來的MAGs的
唯有組裝出的MAGs越完整,未知genome length造成的物種豐度偏差才會越少
如果能組裝出所有metagenome中所有物種的genome,自然就能得知真實的genome length了
忘了說,目前的genome length也只是參考reference genome得來的,跟實際的genome length一定有些許偏差
但這也是沒辦法中的辦法了,有做總比沒做好
計算abundance是做metagenome研究最重要的環節也是最難解的問題了
簡而言之存在越多unclassified物種,偏差就會越多,可怕的是我們並無法得知未知物種到底有多少
除非把整個metagenome所有物種的genome都組裝出來
但是目前組裝出的MAGs幾乎都只是完整genomes的碎片而已,尤其是用NGS reads
難道不能提高定序通量組裝出所有genomes嗎?
可以,但NGS先天存在劣勢導致難度很高,原因網路上查一下就知道了
而Nanopore和Pacbio雖然不時都會發出新聞稿說他們的技術可以組裝出幾乎所有的genomes了
但問題是他們把通量提高到超高,基本上無法作為商用
目前只能用於宣傳而已
Gene catalog
之前都沒看過這名詞,原來只是把相似的gene sequence做clustering而已
統整出metagenome含有的基因
但跟上面談abundance的問題一樣
MAGs絕對不是完整的,所以預測的基因總數絕對是低估的
但還是有做總比沒做好
發現一個滿有趣的資料庫 - Global Microbial Gene Catalog (GMGC)
這團隊把主流幾個採檢來源的metagenome的gene catalog收錄成資料庫
再來是我想到的問題
即使binning過MAGs,但長度還是不會太長,可能導致有些基因是只有組裝出一部分而已(斷頭斷尾)
這些殘缺的基因是否會一起加入clustering,可能有組裝出的部分相似度高,但沒組出來的部分是低的,導致偽陽性
從他們的方法描述是只有去除掉太短的基因而已(<100 bp)
但如果定義很嚴苛一定要有完整的start codon和stop codon的話大概會沒剩多少基因吧
Horizontal gene transfer (HGT)
這種分析方法原來廣泛應用在探討抗藥基因在微生物之間轉移的狀況
簡單來說就是看某個基因是不是有出現在多個分類階層之間,來判斷是不是有HGT的現象發生
MetaCHIP就只是挑出出現在多個物種之間同樣的基因然後再看這是不是抗藥基因
Song, W., Wemheuer, B., Zhang, S. et al. MetaCHIP: community-level horizontal gene transfer identification through the combination of best-match and phylogenetic approaches. Microbiome 7, 36 (2019). https://doi.org/10.1186/s40168-019-0649-y
Sevillya, G., Adato, O. & Snir, S. Detecting horizontal gene transfer: a probabilistic approach. BMC Genomics 21 (Suppl 1), 106 (2020). https://doi.org/10.1186/s12864-019-6395-5
chicken gut metagenome真的很在意抗藥基因
這4篇即使研究目的不是為了探討腸道菌相的抗藥性,還是都會分析metagenome中的抗藥基因豐度和種類
可見抗生素對於經濟動物的影響多深遠,或者只是盲從(X)
回到資料分析的角度,理論上現在最完善且一直有在維護的抗藥基因資料庫非CARD莫屬了
但這4篇居然只有1篇是使用CARD
MEGARes 2.0甚至之前都沒看過,發表該資料庫的文獻是說新增一下antimicrobial compounds, including biocides and metals
就等時間驗證兩個資料庫吧
還有一點他們使用的比對軟體都不是KMA (Clausen et. al., 2018),CARD和ResFinder的開發團隊都是推薦使用KMA來比對他們的資料庫
因為同gene family的抗藥基因之間相似度很高,因而難以判斷reads實際是屬於哪個基因的問題
KMA就是為了解決此問題開發出的序列比對演算法
CARD團隊開發的抗藥基因預測工具RGI就有介紹為何他們推薦使用KMA
https://github.com/arpcard/rgi#analyzing-metagenomic-reads-a-k-a-rgi-bwt
Novel species determination
(Feng et. al., 2021)以相似度(ANI)作為篩選條件將符合的MAGs定義為全新的strain, species或是genus
從他的分析方法頗複雜來看,看得出很努力要盡量要將MAGs和reads都能夠分類taxonomy
但有點大雜燴將各種同類型的工具混在一起使用實在不太好
解釋起來會很困難,頂多就結果好看一點吧
但他們有嘗試要去研究全新(unclassified)MAGs這點倒是其他3篇沒做的
雖然定義完之後頂多只能了解這個採檢來源的metagenome是否存在很多全新物種而已
而且有可能多個MAGs都是來自於同個genome,或是都是不同genomes
這點一樣是無法辨識
頂多可以研究這些未知MAGs含有哪些基因
Conclusion
目前metagenome研究受限於定序技術尚有侷限,無法將所有的MAGs都組裝起來
只能看到部分的物種,但unclassified MAGs也是很珍貴的,
它們會是無法分類的,表示不在已知的物種資料庫中,至少能夠知道他們部分的基因功能
也因為這些unclassified MAGs,使得現行的abundance計量一定都會有偏差
而且還很難估計影響程度,不是忽略它們就是只能硬著頭皮做下去

